SIM-NANOPORE
Programul
Capacităţi / Modulul III - Cooperarea bilaterală 462
/ 18.02.2011
Tip de proiect: Bilateral
Ţari partenere: România - Coreea de Sud
Perioada de colaborare: Martie 2011 -
decembrie 2011
PARTENERI
|
Coordonatori: |
|
|
Dr. Viorel Chihaia Institute of Physical Chemistry “Ilie
Murgulescu”, Romanian Academy Splaiul Independentei 202, 060021, Bucharest Phone (+40) 021-316-7912 e-mail: vchihaia@icf.ro |
Prof. SUH, Soong-Hyuck Keimyung University, Department of Chemical
Engineering #1,000 Shindang-dong,
Dalseo-ku, Daegu, 704-701, Republic of Korea e-mail: shsuh@kmu.ac.kr |
|
Colective: |
|
|
Dr. Popa Vlad |
Prof. Jae-Young Bae |
|
Dr. Munteanu Gabriel |
Prof. Kwan-Kyu Park |
|
Dr. Stanica Nicolae |
Prof. Young_A Son |
|
Dr. Rares Scurtu |
Prof. Tae-Young Kim |
|
Dr. Dascalu Myrella Izabella |
|
Stiinta
este
probabil unul dintre primele domenii globalizate peste intreg globul
pamantesc.
Indiferent de nationalitate, cercetatorii pot lucra impreuna cu succes
bazabdu-se pe interesele lor comune de cercetare si pe capacitatile si
cunostintele lor complementare. Asa numitele programe de mobilitate pot
fi un
bun exemplu. Obiectivul general al prezentului proiect este acela de a
obtine
sprijinul financiar pentru dezvoltarea unei retele efective de
cercetare intre
Romania – Corea. Noi credem ca viziunile larga pentru construirea unei
retele
bilaterale de cercetare ar putea fi una din principalele verigi cheie
pentru
succesul programului de cercetare KICOS-ANCS.
Spre
deosebire de alte grupuri, echipele noastre din Universitatea Keimyung,
Corea
de Sud si Institutul de Chimie Fizica “Ilie Mugulescu”, Romania au o
experienta
indelungata de colaborare. De fapt, cele doua grupuri de cercetare
conduse de
Prof Suh si Dr. Chihaia si-au inceput activitatile de colaborare,
schimb de
idei si sprijin logistic de-a lungu unui deceniu, din 1999. In tot
acest timp
am dezvoltat proiecte de cercetare intercorelate, sprijinindu-ne
reciproc prin
baza materiala (in special de la Universitatea Keimyung) si
cunostintele si
abilitatile complementare ale celor doua grupuri. Un rezultat de seama
a acestei
conlucrari este realizarea clusterului de calculatoare de tip Blade
dezvoltat
la Institutul de Chimie Fizica, in cadrul proiectului CAPACITATI PNI-II
84/2007
realizat in totalitate cu suportul financiar al ANCS (2000000lei).
Prof. Suh a
incurajat si ajutat cu cunostintele sale despre centre High
Performance
Computationsdezvoltate pentru modelare si simulare in stiinta
materialelor.
Mai
mult, incepand cu 2005, noi am initiat si functionat ca membrii
reprezentativi
ai comitetului de organizare a Korea/Romania
Joint Workshop: Molecular Science and Engineering, care este tinut in fiecare an impar si
respectiv par in Corea si
respectiv in Romania. Acest
workshop asigura un forum efectiv bilateral intre Romania si Corea in
domenii
ale chimie, fizici si stiintei moleculare precum si in domenii
ingineresti
inrudite. Mai mult decat colaborarile Corea-Romania, noi dezvoltam
proiecte de
cercetare in cooperare cu alte tari din EU printre care Spania,
Germania,
Ungaria si Bulgaria. Noi ne propunem de asemenea sa creem proiecte de
cercetare
in cadrul EU-FP7 prin perspectiva parteneriatului de cercetare ce se va
realiza
prin intermediul prezentului program.
DESCRIEREA PROIECTULUI
Materialele
moi (soft materials) sunt materiale cu stari fizice caracterizate prin
valori ale
energiei in scala energiei termice corespunzatoare temperaturii
camerei. In
ciuda formelor variate ale acestor materiale (lichide, coloizi,
surfactanti,
polimeri, spume, geluri, microemulsii, materiale granulare, membrane si
unele
matriale biologice) multe din proprietatile lor au origini
fizico-chimice
comune, cum ar fi un numar mare de grade de libertate, interactii slabe
intre
elementele structurale si o balanta echilibrata intre contributiile
entropiei
si entalpiei la energia lor libera. Astfel de sisteme au o structura
complexa
dificil de descris direct prin constituentii lor atomici sau
moleculari.
Particulele constituente pot contine mii si chiar milioane de atomi,
interconectati intr-un mod complicat. Aceste sisteme se
auto-organizeaza in
structuri fizice mesoscopice (cu dimensiuni tipice intre 1 nm-1mm)
care sunt mai
mari de cat scala microscopica dar mult mai mica decat scala
macroscopica a
materialului. Aceste meso-obiecte au o forma variata precum siruri
lineare,
ramificate de tip stea sau dendritice si de copolimeri. Au o varietate
de
functii precum stabilizatori sterici, aditivi si dispersivi.
Flexibilitatea lor
poate influenta structura si comporatarea solutiilor polimerice prin
schimbarea
calitatii solventului.
Proprietatile
si
interactiile acestor stucturi mesoscopice pot determina comportarea
macroscopica a materialului. Datorita flexibilitatii lor in comparatie
cu
contracandidatii lor atomici permit manipularea particulelor
constituente la
nivel molecular. Astfel se poate obtine o varietate foarte mare de
arhitecturi,
cu comportari variate. Materialele moi sunt caracterizate prin
interactii slabe
intre componentii moleculari sau supramoleculari si au deobicei o forma
amorfa
sau se pot auto-ansambla din stari licide. Adesea, aceste materiale
prezinta
multe nivele de complexitate cu structuri supramoleculare ierarhizate
ce pot fi
concurente si in stari departe de echilibru.
Adesea
suntem
interesati de aranjamentul structural, reologia vasco-elastica si/sau
comportarea mecanica a acestor materiale. Aceste proprietati pot avea
caracteristici diferite atunci cand materialele moi sunt intoduse in
materiale
cu cavitati sau pori de dimensiuni nano. Atractiile de raza scurta apar
a juca
un rol minor in comportarea macroscopica a acestor sisteme.
Tema
fundamentala a
acestui proiect este dezvoltarea de metode de investigare specifice
simularii
moleculare si de tehnici de caracterizare a sistemelor moi confinate in
materiale poroase nanostructurate. Noi folosim diferite metode de
simulare
moleculara impreuna cu meodele de caracterizare termodinamice si
statistice
pentru determinarea detaliata a proprietatilor molecuare, care foarte
adesea
sunt dificil sau imposibil de masurat experimental. Compararea cu
datele
experimentale poate fi folosita in investigarea modelelor sistemelor
nanoporoase. Scopul final al simularii moleculare de a realiza
experimente
(numerice) ce nu pot fi efectuate in laboratorul real. Cercetarea in
acesta
directie poate deschide cai de design efectiv si de imbunatatire a
producerii de
materiale nanoporoase si nanocomposite necesare dezvoltarii de
nanotehnologii
avansate.
Pentru
diferitele
arhitecturi de particule mesoscopice interactia dintre coordinatele
alese
corespunzator sunt foarte slabe (ultrasoft), adica acestea sunt finite
sau
diverg foarte lent la separatii zero. Drept consecinta, fazele fluide
au
caracteristeci neuzuale precum comportari anormale ale functiei de
corelatie
interparticule si a campului mediu precum si comportarile
termodinamice.
Potentialele ultrasoft si de legatura reprezinta modele foarte utile in
caracterizarea interactiei efective de tip two-body in sisteme
coloidale si de
polimeri cum ar fi polimerii de tip lant sau stea in solventi. Cel mai
simplu
potential legat include modelul sferei penetrabile ce poate fi vazut ca
un caz
limita a modelului generalizat Gausian de tip core. Acest potential
penetrabil
de interactie, calitativ similar unei functii step nu neaparat
divergenta in
centrul molecular, se poate folosi pentru micele si fluide complexe
confinate in
diferite sisteme nanoporoase. Lipsa limitelor potentialului penetrabil
conduce
mai degraba la comportari de faze exotice ce se intalneste in materiale
moi cum
ar fi topirea si clusterizarea in stari legate. Ecuatiile neliniare de
integrare, in particular aproximatiile Percus-Yevick si a lanturilor
supralegate, nu descriu la un nivel satisfacator structura acestor
fluide
penetrabile, mai ales pentru temperaturi joase si la concentratii mari.
Ca
un instrument
intermediar intre teorie si experiment, simularea moleculara poate
caracteriza
proprietatile de echilibru si de transport pentru fluide penetrabile
constanse
in nanopori, fiind in acelasi timp un instrument de nepretuit in
judecarea
calitatii aproximatiilor bazate pe mecanica statistica si pe
termodinamica. Pe
langa folosirea acestui instrument, ne vom concentra pe dezvoltarea de
algoritmi si metode de caracterizare a sistemelor investigate
bazandu-ne pe
conectivitatea interatomica, precum clusterizarea, stratificarea,
topologiile
ciclurilor, formarea canalelor si a cavitatilor. In afara de aceste
criterii
structurale, o alta atentie metodologica va fi data dezvoltarii
algoritmilor de
caracterizare a proprietatilor dinamice. O atentie speciala se va
acorda
caracterizarii topologice a sistemelor mari si/sau periodice legate
prin
legaturi de hidrogen.
Studierea
prin simulari de dinamica moleculara si Monte Carlo a proprietatilor de
echilibru si transport a materiei moi condensate va fi importanta
pentru
aplicatii si pentru design-ul de materiale nanoporoase. In plus fata de
aceste
calcule clasice, vom parametriza potentialele model specifice
sistemelor
nanoporoase prin compararea cu date experimentale si cu date rezultate
din
calcule cuanto-chimice semiempirice si ab-initio.
Deoarece
simularile
si modelarile asistate de calculator nu sunt constranse de limitele
curente ale
capacitatii noastre de a prepara nanostructuri specifice, acestea se
constitue
in instrumente ideale pentru explorarea si examinarea potentialalelor
structuri
de dimensiuni nanometrice, ce reprezinta principalele obiective al
nanotehnologiilor. Eforturi deosebite se fac in ambele tari in
dezvoltarea de
metode de investigatii si de tehnologii ale materialelor moi confinate
in
sisteme nanoporoase.
Interesele
de
cercetare ale Prof. Soong-Hyuck Suh sunt asociate teoriilor mecanicii
statistice si simularilor moleculare asistate de calculator, in special
a
proprietatilor de echilibru si transport a fluidelor si amestecurilor
confinate
in sisteme model nanoporoase. Contributia sa in acest proiect este o
continuare
a muncii sale curente de cercetare pe modele de sisteme nanoporoase la
nivel
atomic si molecular. A implementat potentiale ultrasoft si de legatura
in codul
de simulare dezvoltat in laboratorul sau, permitand utilizatorilor
acestuia sa
creasca complexitatea sistemelor ce pot fi modelate in domeniile
nanomaterialelor, cristalelor moleculare, polimerilor si chimiei
organice.
Programul sau contine o metoda de investigare de tip coarse-grained ce
permite
considerarea in cadrul acelorasi calcule combiantii de materiale de tip
soft si
condensate.
Dr.
Chihaia are
experienta in simularile moleculare bazate pe metode cuantice si
clasice de
chimie cuantica si fizica solidului si metode de tip embed pentru
diferite
fenomene si sisteme atomice si moleculare extinse, precum ar fi,
adsorbtia si
difuzia moleculelor si a clusterilor mici pe diferite suprafete,
caracterizarea
structurii si a comporatrilor dinamice a compusilor de tip canale si
cavitati
(nanotuburi de carbon, siliciu poros, clusteri de apa de tip buckyball
si
hidratilor clatrat). A dezvoltat diversi algoritmi de calcul. Studiile
sale in
aceasta directie include dinamica corpului rigid, rutine eficiente,
bazate pe
conectivitatea atomilor, de determinare a ciclurilor si cavitatilor cu
dimensiuni arbitrare, proceduri de caracterizare a topologiei
legaturilot de
hydrogen si construirea autamata a suprafetei de energie potentiale
pentru
fenomene de adsorptie si difuzie. Recent a initiat si dezvoltat pentru
prima
data in Romania un cluster de tip Blade pentru calcule de inalta
performanta
pentru simulare in stiinta materialelor la nivel multiscala. In
proiectarea si realizarea
acestui cluster a avut suportul real al Prof. Suh.
Cele
doua grupuri
se completeaza reciproc prin abilitatilelor si dotarile lor materiale.
Realizarile lor comune demonstreaza eficienta acestei colaborari.
REZULTATE
Pentru a testa
instalarea
codurilor am ales ca sisteme de lucru materialele moi (soft materials)
ce sunt
materiale cu stari fizice caracterizate prin energii cu valori
apropiate de
energia termica corespunzatoare temperaturii camerei. In ciuda formelor
variate
ale acestor materiale (lichide, coloizi, surfactanti, polimeri, spume,
geluri,
microemulsii, materiale granulare, membrane si unele matriale
biologice) multe
din proprietatile lor au origini fizico-chimice comune, cum ar fi un
numar mare
de grade de libertate, interactii slabe intre elementele structurale si
o
balanta delicata intre contributiile entropiei si entalpiei la energia
lor
libera. Astfel de sisteme au o structura complexa dificil de descris
direct
prin constituentii lor atomici sau moleculari. Particulele constituente
pot
contine mii si chiar milioane de atomi, interconectati intr-un mod
complicat.
Aceste sisteme se auto-organizeaza in structuri fizice mesoscopice (cu
dimensiuni tipice intre 1 nm-1µm) care sunt mai mari de cat scala
microscopica
dar mult mai mica decat scala macroscopica a materialului. Aceste
meso-obiecte
au forme variate precum siruri lineare, ramificate de tip stea,
dendritice si
copolimeri. Au o varietate de functii precum stabilizatori sterici,
aditivi si
dispersivi.
Flexibilitatea lor
poate
influenta structura si comporatarea solutiilor polimerice prin
schimbarea
calitatii solventului.
Proprietatile
si
interactiile acestor stucturi mesoscopice pot determina comportarea
macroscopica a materialului. Flexibilitatea lor foarte mare constitue
un
avantaj al lor in comparatie cu contracandidatii lor atomici deoarece
permit
manipularea particulelor constituente la nivel molecular. Astfel se
poate
obtine o varietate foarte mare de arhitecturi, cu comportari variate.
Putem enumera
cateva exemple de
materiale moi: lichide, coloizi, polimeri, spume, geluri, materiale
granulare
dar si de sisteme mai complexe precum ar fi suspensii bacteriene şi
polimeri biologici. De obicei materialele moi au interactii slabe intre
componentii moleculari sau supramoleculari si au de obicei o forma
amorfa sau
se pot auto-ansambla din stari licide. Adesea, aceste materiale
prezinta multe
nivele de complexitate cu structuri supramoleculare ierarhizate ce pot
fi
concurente si in stari departe de echilibru. De mare interes pentru
colectivele
noastre sunt sistemele surfactant, in particular cele formate de
sulfatul de
sodiu. Sodiu dodecil sulfatul (SDS - Sodium Dodecyl Sulfate) este un
detergent
anionic ce denatureaza proteinele si care confera o incarcatura
electrica negativa
complexului SDS-proteina, ceea ce ne permite controlul migrarii
proteinelor
functie de masa moleculara a acestora si de incarcarea lor electrica.
Este
foarte util in separarea nano-obiectelor precum grafene, nanotuburi sau
fulerene si in controlul formarii sistemelor nano- si meso-poroase. Mai
jos
prezentam rezultatele obtinute in modelarea catorva sisteme in
componenta
carora intra solutii apa – SDS.
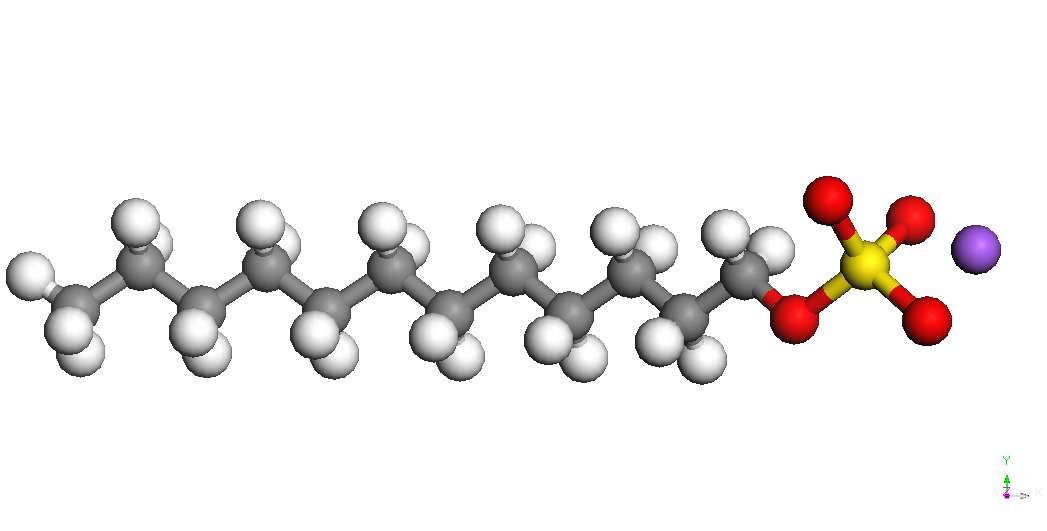
Sodiu dodecil sulfat
-
SDS
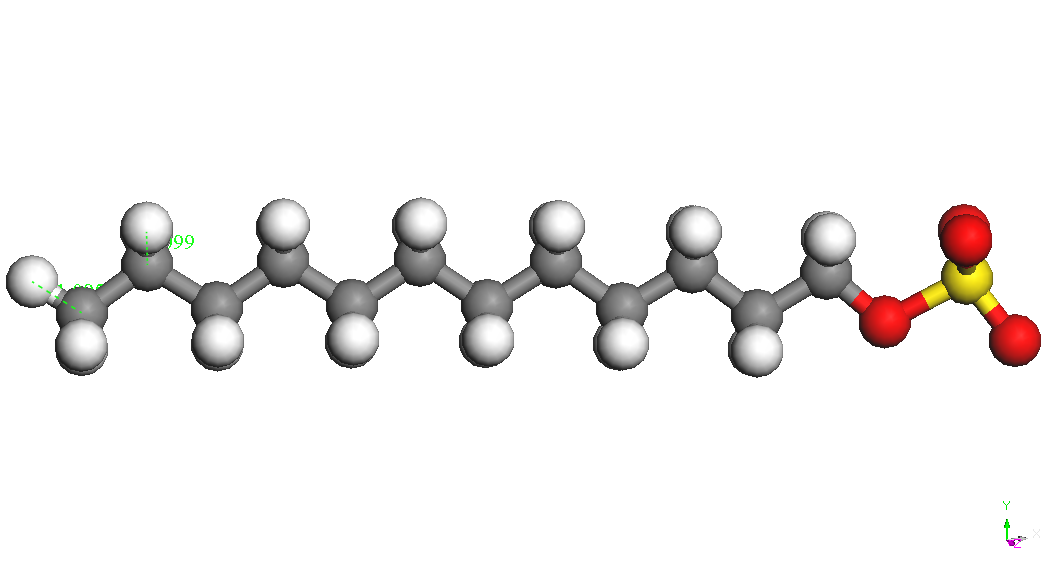
Sodiu dodecil sulfatul este
un compus organic cu formula NaC12H25SO4
sau
CH3(CH2)11OSO3Na,
fiind format
dintr-un lant de 12 atomi de carbon atasat unui grup sulfat OSO3
prin intermediul unuia dintre oxigeni. Este un agent tensioactiv
anionic.
BE(Na)
= E(CH3(CH2)11OSO3Na)
– [E(CH3(CH2)11OSO3-)
+
E(Na+)]
unde E(CH3(CH2)11OSO3Na)
este energia intregului sistem, E(CH3(CH2)11OSO3-)
este energia lantului CH3(CH2)11OSO3-
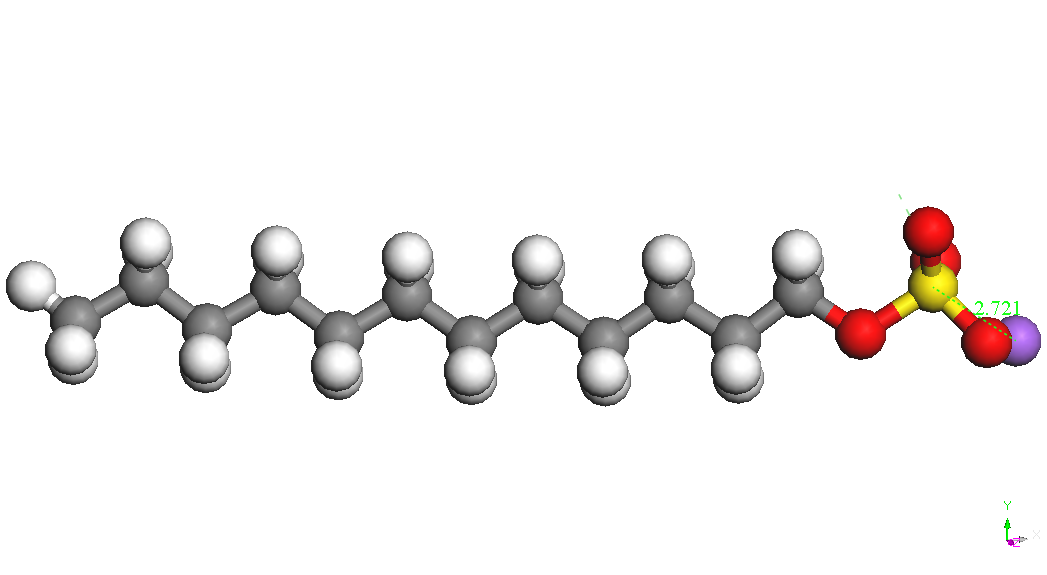
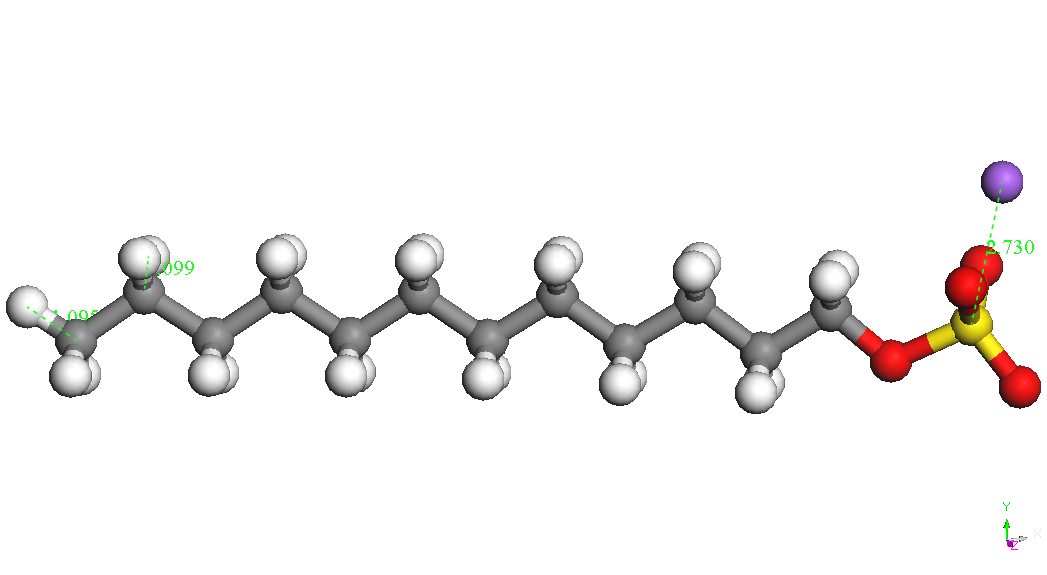
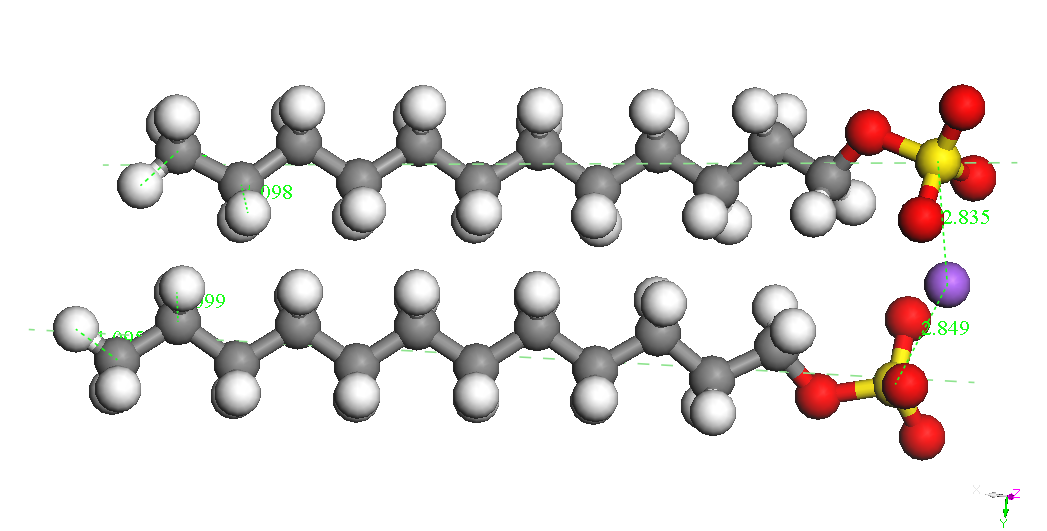
iar E(Na+) este energia ionului de sodiu izolat (104.015 kcal/mol). Cel mai stabil conformer al CH3(CH2)11OSO3-
este prezentat in Tabelul 1. Ionul de sodiu se leaga de gruparea sulfat
in
diverse pozitii la o distanta S-Na de aproximativ 2.72 A. Cel mai
stabil
conformer este cel denumit CH3(CH2)11OSO3Na,
Na – down in Tabelul 1.
Tabelul 1. Diverse structuri
asociate sistemului
SDS si energiile lor de legatura (BE).
|
|
[CH3(CH2)11OSO3]- BE = -4322.832kcal/mol |
|
|
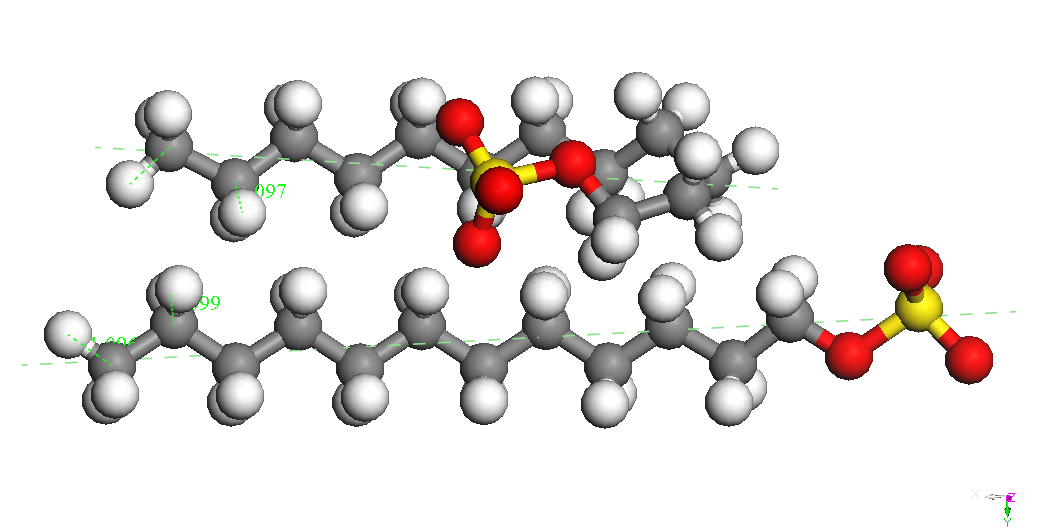
CH3(CH2)11OSO3Na,
Na –
down (twisted) BE =
-4337.834 kcal/mol BE(Na) =
-74.177 kcal/mol |
|
|
CH3(CH2)11OSO3Na
, Na –
up BE = -4336.335 kcal/mol BE(Na) =
-72.678 kcal/mol |
|
|
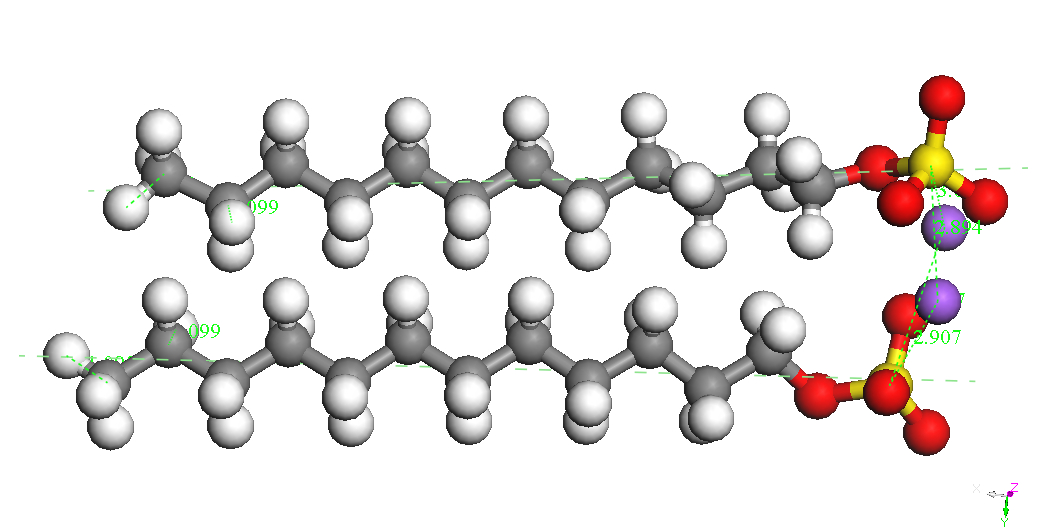
Dimer Head-Head Parallel 1 BE = -8743.650 kcal/mol |
|
|
Dimer Head-Head Parallel 2 BE = -8730.682kcal/mol |
|
|
Dimer Head-Head Along BE = -8715.747kcal/mol |
|
|
Dimer Head-Head perpendicular BE = -8697.493kcal/mol |
|
|
Dimer-Na Head-Head Parallel 2 BE = -8744.893kcal/mol |
|
|
Dimer-Na Head-Head Parallel 1 BE = -8735.920 kcal/mol |
|
|
Dimer-Na Teil-Head Parallel BE = -8721.021 kcal/mol |
|
|
Dimer-2Na Head-Head Parallel 2 BE = -8670.233kcal/mol |
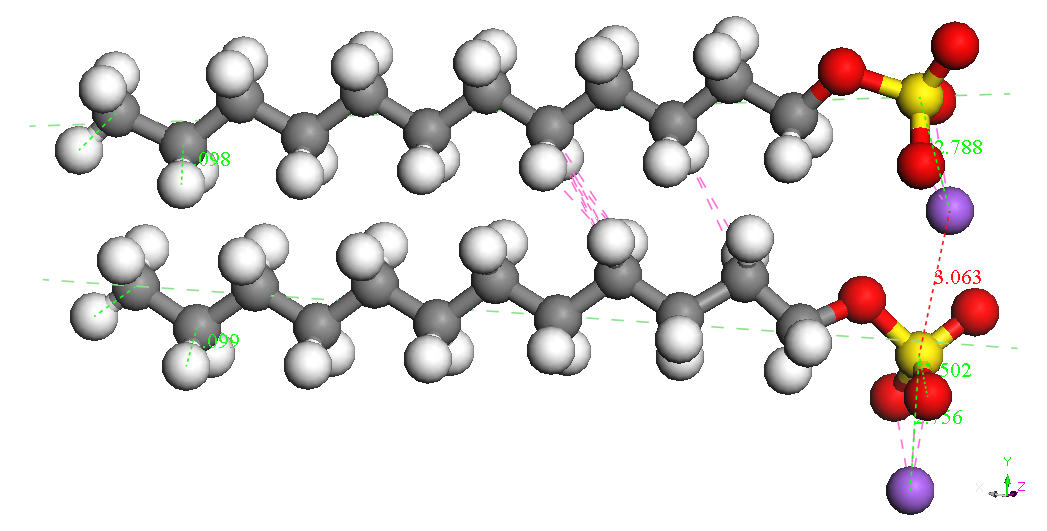
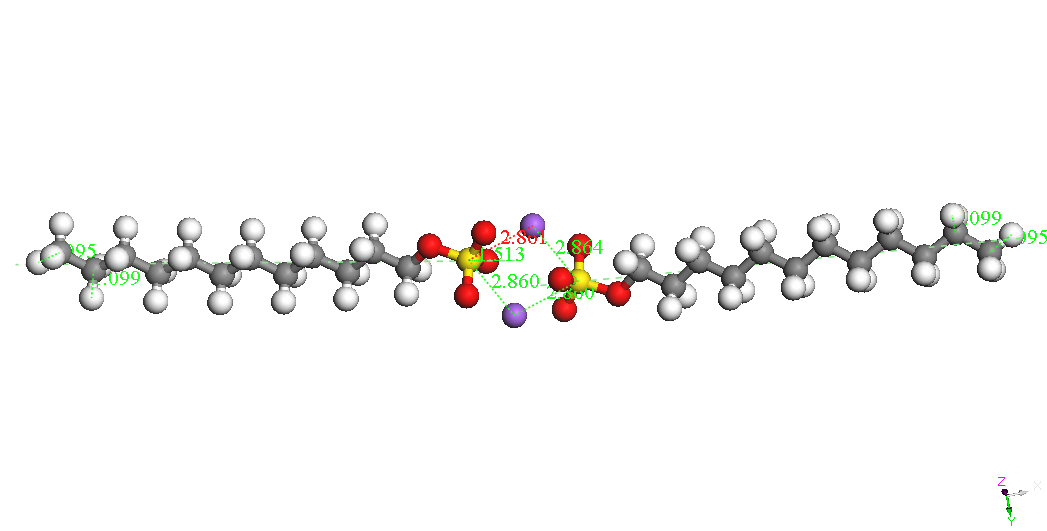
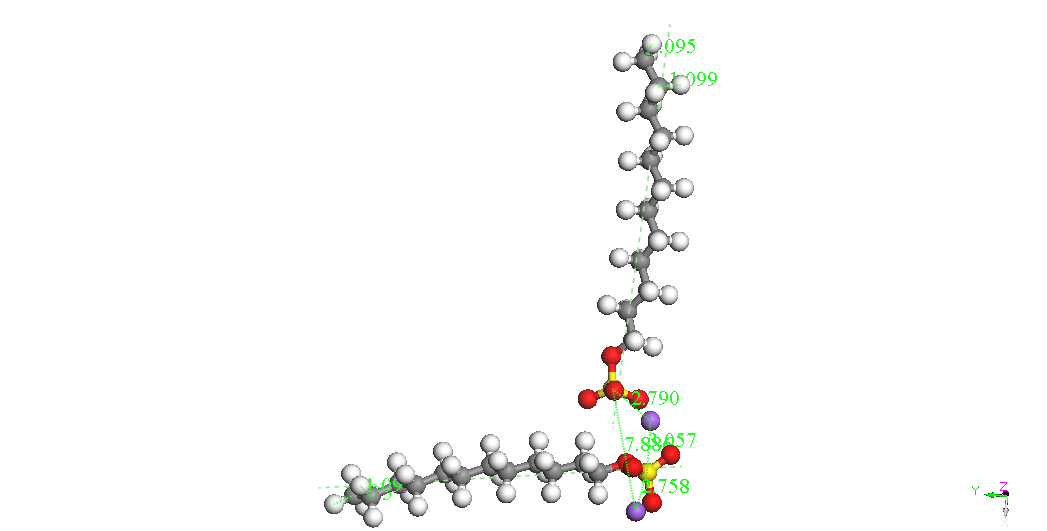
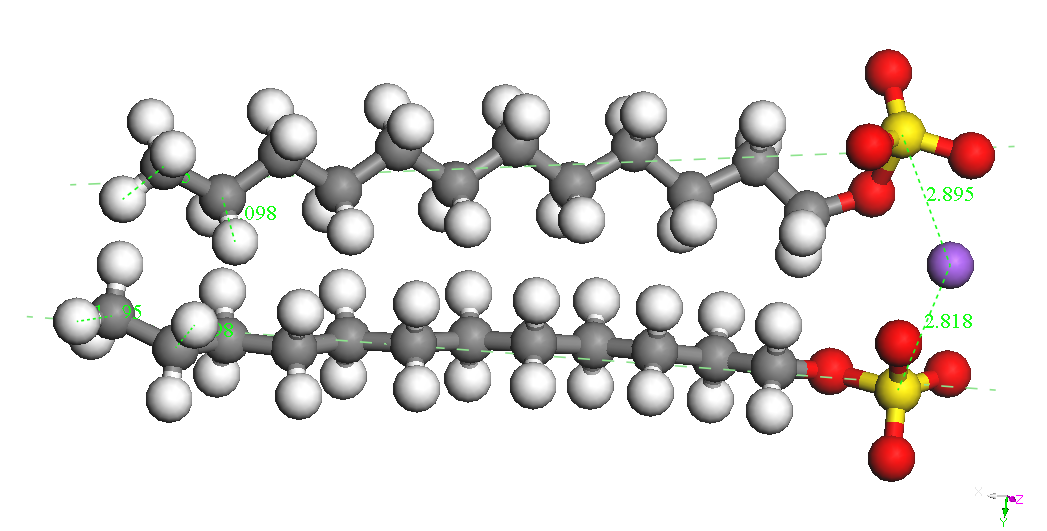
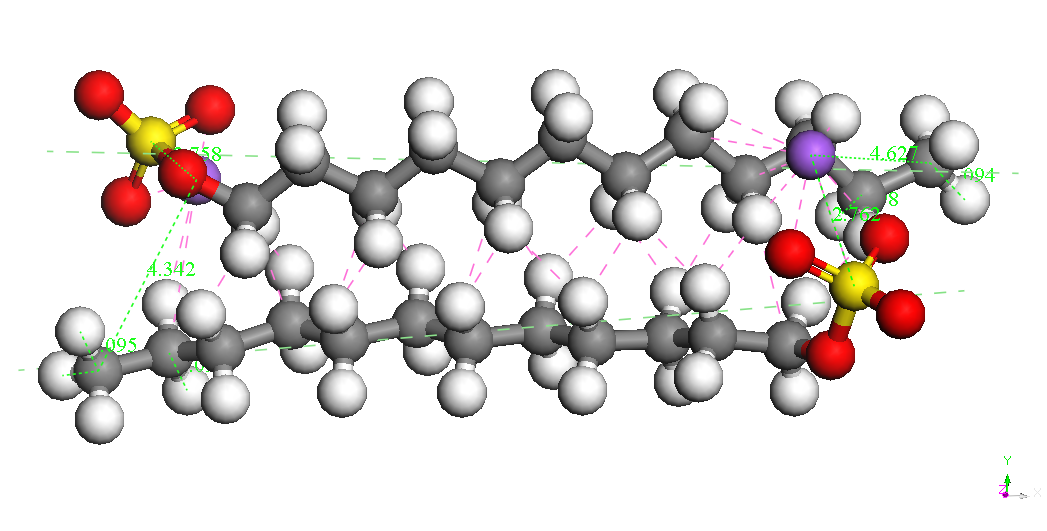
In cazul dimerilor izolati se
poate constata ca
cel mai stabil izomer este cel denumit Dimer Head-Head Parallel 1,
pentru cele
doua molecule SDS sunt paralele, cu aceeasi orientare si pentru care
cei doi
ioni Na+ sunt
asezati de-o parte si de alta a celor
doua grupari sulfat. Este posibil ca imersia sistemului in apa sa
modifice
polarizarea moleculelor SDS ceea ce ar afecta stabilitatea izomerilor
dar nu ne
asteptam sa modifice asezarea paralela si orientarea celor doua
molecule. Un
studiu abinitio ar fi foarte costisitor datorita numarului mare de
molecule si
de configuratii ce ar trebui investigate. Faptul ca moleculele SDS
formeaza
micele in apa pentru concentratii mai mari decat o valoare critica
sugereaza un
aranjament paralel preferentiat al moleculelor SDS.
Parametrizarea campurilor de forta
Coarse
Grain pentru sistemele SDS, apa si forme alotropice ale carbonului
Suprafata de energie
potentiala in cazul campurilor d eforta de tip coarse grain este mult
mai
neteda decat a celor de tip all-atoms (AA). Principala cerinta a
acestor
campuri este aceea de a reproduce miscarea de ansamblu a grupurilor
atomice ce
sunt reprezentate prin pseudoparticulele beads. De obicei potentialele
CG sunt
parametrizate astfel incat sa reproduca functia de distributie radiala
a
diferitor perechi de elemente din sistem sau prin inversarea ecuatiei
Boltzann
din simulari de dinamica moleculara AA. De aceea aceste potentiale
depind de
parametrii de rulare a simularilor de dinamica moleculara all-atoms
printre
care pasul de timp folosit pentru integrarea ecuatiei de miscare si
temperatura
sistemului. Pasul de timp in simularile CGMD poate fi considerat de 5
pana la
20 de ori mai mare decat cel folosit in cazul simularilor AA, permitand
astfel
studiul fenomenelor ce se petrec la scale de timp mai mari. Totodata se
reduce
numarul de particule intre care se calculeaza fortele de interactie si
a caror
ecuatie de miscare se integreaza. Astfel efortul de calcul este mult
redus,
permitand studiul unor sisteme mai mari si timpi mai mari. Pe de alta
parte,
fortele si potentialele CG au forme matematice mai simple, in general
de
distanta scurta, deoarece potentialele sunt de obicei trunchiate.
In acest studiu am preluat
din literatura parametrii campului de forte CG de tip Martini 2.0 [S.J.
Marrink, H.J. Risselada, S. Yefimov, D.T. Tielman and A.H. de Vries,
J.Phys.Chem.B, 111, 2007, 7812] si i-am folosit pentru sistemele de
inters dupa
o verificare a consistentei parametrilor. Astfel am efectuat calcule de
dinamica moleculara CG pentru sistemele prezentate in Tabelul 2. In
potentialele Martini patru molecule de apa sunt reprezentate printr-o
singura
pseudo-particula P4; in molecula SDS un fragment de patru carboni si 4
sau 5
hidrogeni este reprezentat printr-o particula C2, gruparea sulfat SO4- este reprezentata printr-o particula Qa incarcata
-1, iar ionul de sodiu
printr-o particula Qd incarcata +1; in sistemele de carbon, doi atomi
de carbon
legati sunt reprezentati printr-o particula SC4. Pentru ca sistemele CG
de apa
au tendinta de a ingheta la temperatura camerei, am inlocuit cu
particule BP4
intre 5 si 10 % din particulele P4. Acestea au rolul de a perturba
aranjarea
moleculelor de apa in structuri ordonate si a impiedica ‚‚inghetarea‘‘
sistemului.
Am construit doua sisteme
amorfe de apa si SDS formate dintr-un numar de 20000 molecule in
reprezentarea
AA si sistemele de carbon prezentate in Tabelul 2. Am transformat
sistemele AA
in sisteme echivalente CG prin inlocuirea grupurilor de atomi cu
corspondentii
lor CG. Pentru reducerea tensiunilor interne am optimizat structurile
CG intai
mantinand fixe forma si dimensiunea celulelor dupa care am relaxat
complet
sistemele. S-au efectuat simulari in ansamble termodinamice NVT pentru
o
temperatura de 300 K, mentinuta printr-un termostat Nose cu un
parametru Q=100.
S-au incercat diversi pasi de timpi, cei optimi pentru toate sistemele
fiind de
25 fs, ceea ce reprezinta o crestere a scalei de timp de aproximativ 50
de ori
fata de simularile AA. Sistemele au fost echilibrate pentru 20 ns si
valorile
termodinamice au fost mediate din alte 10 ns. Rezultatele au fost
comparate cu
cele obtinute din simulari AA pentru un pas de timp de 0.5 fs si pentru
o
perioda de echilibrare de 200 ps. S-a constat o reproducere
satisfacatoare a
functiilor de distributie radiale. S-au repetat simularile pentru
ansamble NPT,
in conditiile unei presiuni de 1 atm folosind un barometru Berensen.
Constantele de retea obtinute in simualrile AA sunt reproduse intr-o
marja de
eroare de 5% in calculele CG.
Tabelul 2. Reprezentarea CG a
sistemelor atomice
de interes in prezentul studiu
|
Sistem |
Reprezentare atomistica Elemente constituente – Atomi |
Reprezentare Coarse Grained Elemente constituente – Beads |
|
4 molecule de apa |
|
|
|
SDS |
|
|
|
CNT (12,12) |
|
|
|
Carbon amorf |
|
|



Micele SDS imersate in
apa
Am verificat reproducerea
valorilor densitatii apei pure si a solutiei apa-SDS prin simulari de
dinamica
moleculara CG pentru 20 ns, in conditiile folosirii unui ansamblu
statistic NPT
(T = 300K, P = 1 atm, barostat Berendsen). Densitatea sistemelor
initiale a
fost de 2 g/cm3 si
sau obtinut dupa o perioada de 500 ps
densitatile de echilibru de 1 respectiv 2 g/cm3.
In solutia apa-SDS
moleculele de SDS formeaza aglomeratii in care moleculele sunt
orientate cu
ionii de sodiu in aceesi directie. Din alte studii din literatura
rezulta ca
pentru peste o valoare critica a concentratie SDS, aceste molecule se
aduna in
clustere cvasi-sferice in care moleculele de SDS se aliniaza radial cu
capetele
unde ionii Na+ sunt
prezenti, in afara clusterelor.
Aceste clustere sunt stabile in timp si sunt de fapt micele.
|
Apa densitate de echilibru 1.0 g/cm3 |
|
|
Solutie apa-SDS structura initiala |
|
|
structura finala, densitatea 0.8 g/cm3 |
|



Figura 2.
Sistemele folosite in
studiul reproducerii densitatii apei pure si a solutiei apa-SDS.
|
0
ns |
|
|
50
ns |
|
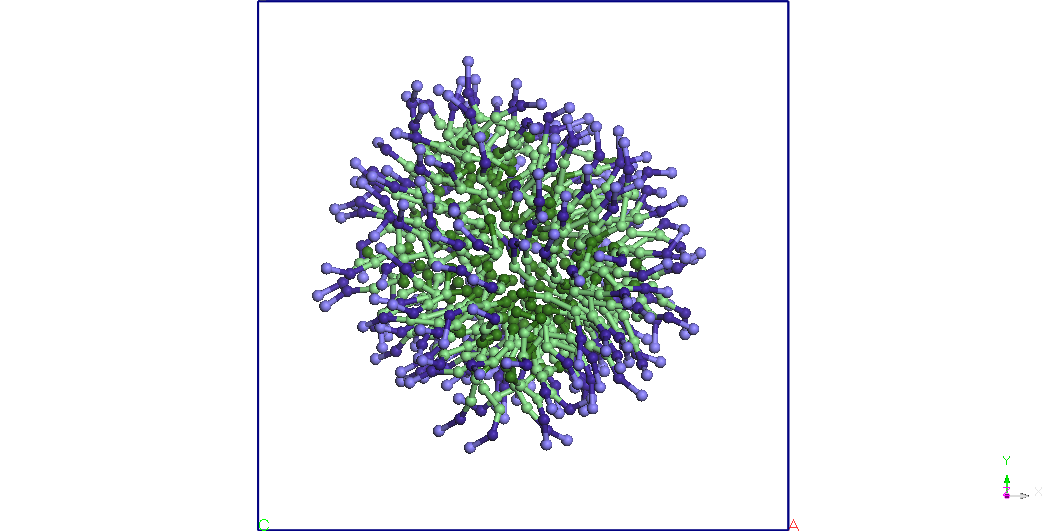
Figura 3.
Structura unei micele
de SDS imersata in apa. Pentriu claritate moleculele de apa nu sunt
reprezentate.
Pentru o concentratie de 2
molecule de sodiu la 3 molecule de apa am format o micela care ramane
stabila
si aproape sferica dupa 50 ns de simulare CG pentru un ansamblu
termodinamic
NVT. T=300K (vezi Figura 3).
Folosind structura
echilibrata a micelei SDS am construit prin dublarea structurii un
dimer
micela-SDS. Distanta initiala dintre centrele de greutate a celor doua
micele
este de 8.46 nm. Am relaxat sistemul astfel format pentru 1ns intr-un
ansamblu
NPT (T=300K, P=1 atm, barostat Berendsen). Se observa ca la sfarsitul
simularii
cele doua micele se apropie la o distanta medie de 5,60 nm (vezi Figura
4).
Cele doua micele isi modifica usor forma, initial sferica. Am continuat
simularea in ansamblul NVT prin fixarea dimensiunii cutiei de simulare
obtinute
in simularea NPT si pastrand celelate conditiile ale simuarii CG
anterioare. Se
observa ca dupa 5ns cele doua micele se alungesc si-si modifica
orientarea,
apropiindu-se la doua capete. Dupa aproximativ 9.5 ns distanta dintre
cele doua
terminatii se reduce si mai mult si apar difuzii de molecule SDS de la
o micela
la alta. Un urma acestor migratii de molecule SDS una dintre cele doua
micele
isi reduce dimensiunile in favoarea celeilalte micele (dupa aproximativ
15 ns).
Dupa 25 ns, nu se mai observa difuzii de molecule SDS intre cele doua
micele.
Cele doua micele isi pastreaza forma si orientarea pana la sfarsitul
celor 40
ns de simulare.
|
|
|
|
|
0
ps / NPT |
100
ps / NPT |
1
ns / NPT |
|
|
|
|
|
2
ns / NVT |
5
ns / NVT |
10
ns / NVT |
|
|
|
|
|
15
ns / NVT |
25
ns / NVT |
40
ns / NVT |
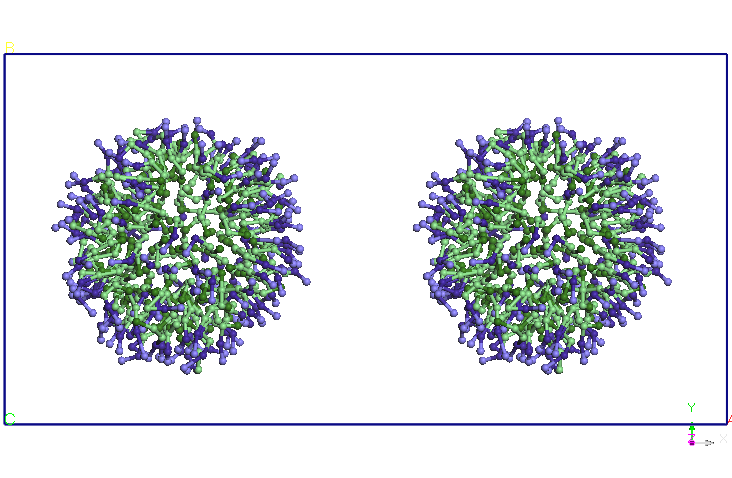
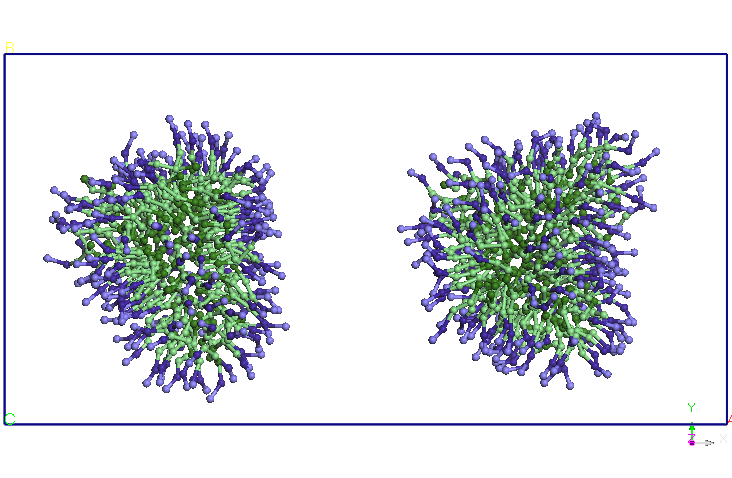
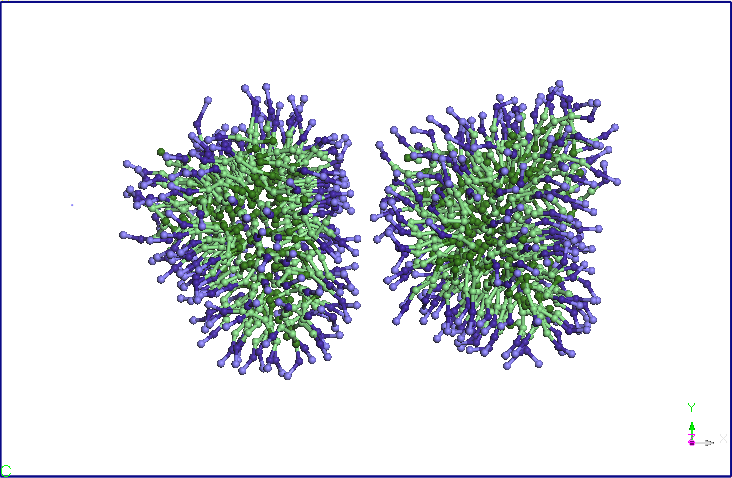
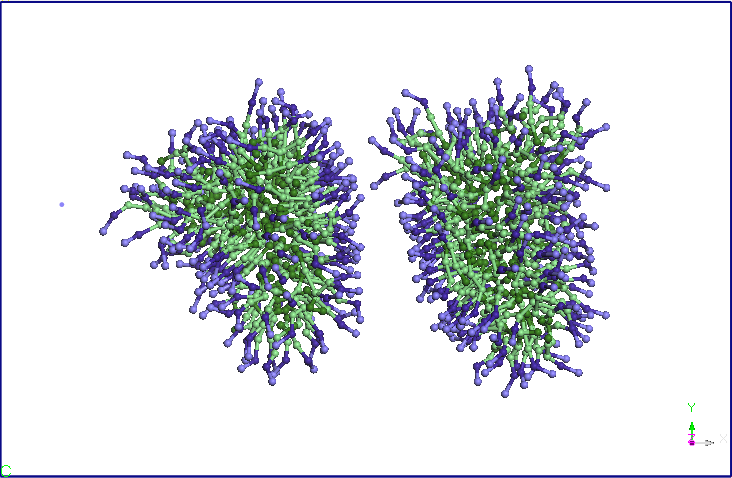
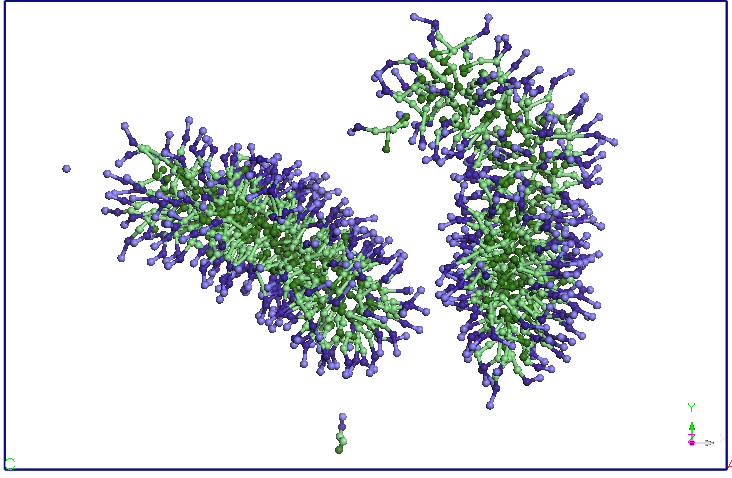
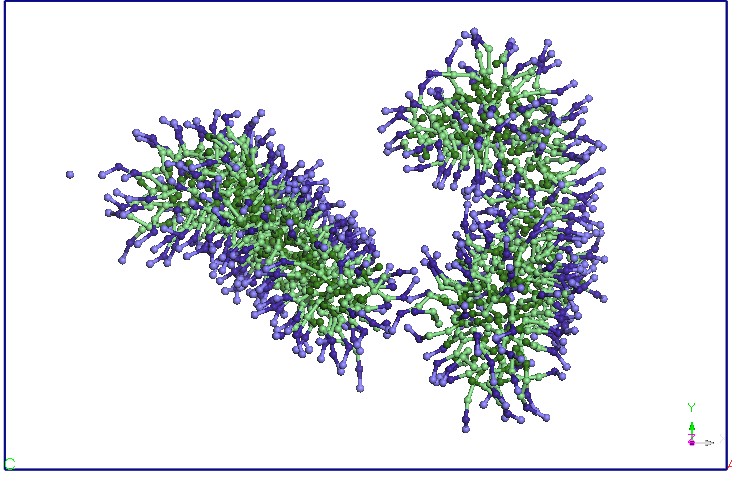
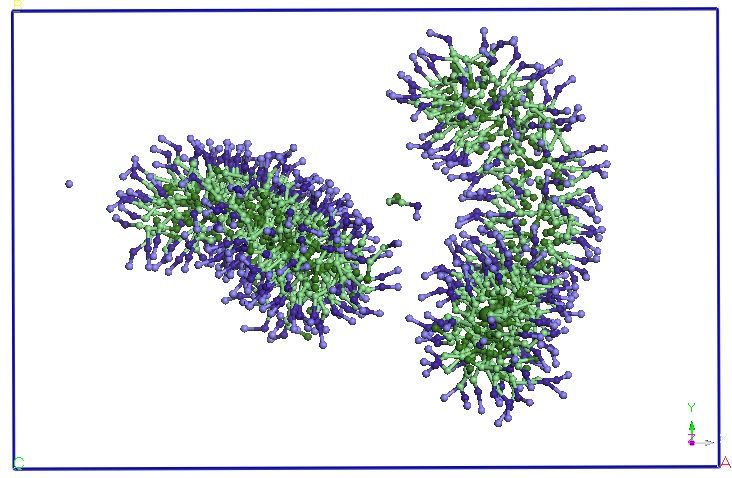
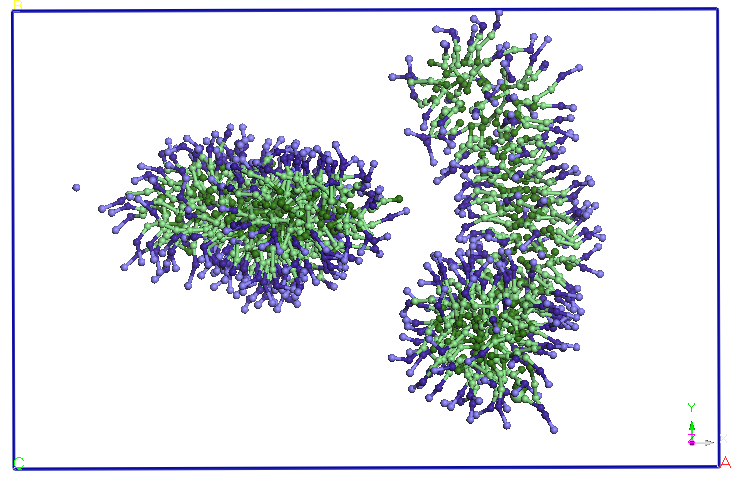
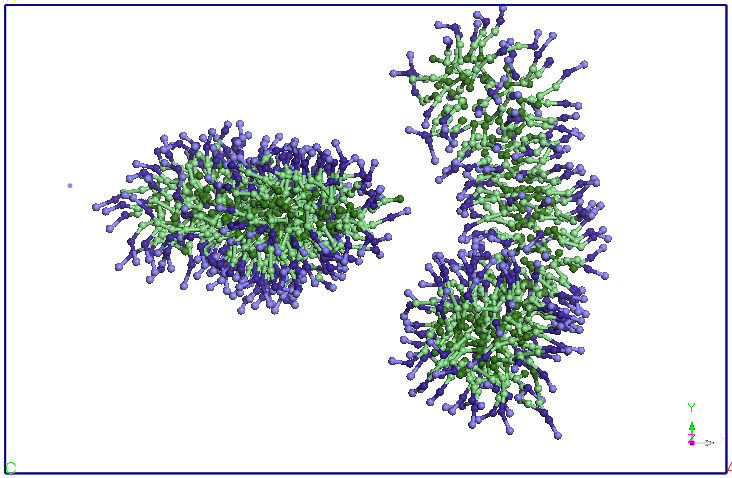
Figura 4. Evolutia
sistemului de
doua micele imersate in apa in simularile de dinamica moleculara in
ansamblele
termodinamice NPT si NVT. Pentriu claritate moleculele de apa nu sunt
reprezentate.
Este important sa mentionam
ca in decursul acestor simulari moleculele de SDS isi pastreaza mai tot
timpul
orienttarea cu capatul hidrofilic (Na+) catre
exterior, catre apa.
Difuzia prin nanosite de carbon amorf
Sitele cu dimensiunea ferestrelor
de ordinul
nanometrilor au o importanta deosebita in selectivitate si producerea
de
sisteme ordonate, jucand rolul de masca in prepararea acestora. Am
ansamblat in
aceasi celula de simulare doua straturi de grafene, un strat de solutie
apa-SDS
si o sita de carbon amorf cu ferestre/pori cu dimensiunea medie de 13 A
(vezi
Figura 5). Cele trei sisteme sunt echilibrate separat prin simulari de
dinamica
moleculara CG la o temperatura de 300 K. Straturile de grafena si
carbon amorf
sunt separate printr-un spatiu gol. Straturile de grafena au rolul de a
separa
solutia apa-SDS si eventualele molecule de apa si SDS ce ar migra prin
ferestrele carbonului poros.
|
|
|
|
|
|
|
|
|
Doua
starturi grafena |
Solutie
apa-SDS |
Nanosita
carbon amorf |




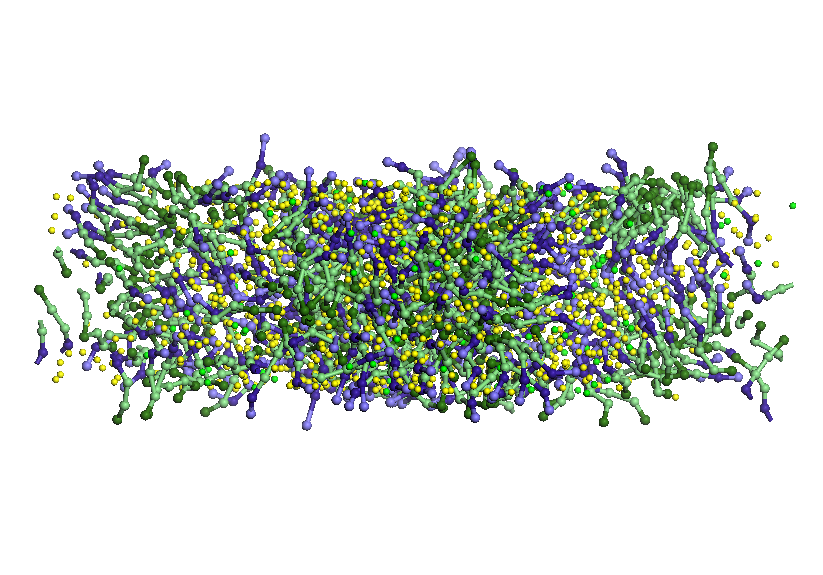

Figura 5.
Imaginile de sus si
frontale a componentelor sistemului investigat.
|
|
|
|
|
0 ns |
0.05 ns |
50 ns |



Figura 6. Evolutia
sistemului in
timpul simularilor de dinamica moleculara, in conditiile impunerii
uniui sistem
poros rigid, pentru pori cu dimensiunea medie de 13 A.
Pentru a vedea rolul jucat
de rigiditatea structurilor de carbon am efectuat doua simulari: i)
mentinand
in pozitii fixe pseud-particulele de Carbon si ii) lasandu-i sa se
deplaseze
liber conform dinamicii Newton. Ambele simulari sund efectuate in
ansamble
termodinamice NVT prin mentinerea fixa a celulei elementare si a
temperaturii
T=300K, controlata cu un termostat Nose cu parametrul Q=100.
In conditiile mentinerii
fixe a pseudo-particulelor de carbon se constata ca in decursul
primelor 50 ps,
solutia apa-SDS se ‚lipeste‘ de carbonul poros si cateva molecule de
apa si SDS
patrund in interiorul ferestrelor (vezi Figura 6) si raman localizate
acolo pe
intreaga durata a simularii (50ns). Numai cateva molecule de apa ajung
in zona
libera, nici o molecula SDS neintrand insa in aceasta zona.
|
|
|
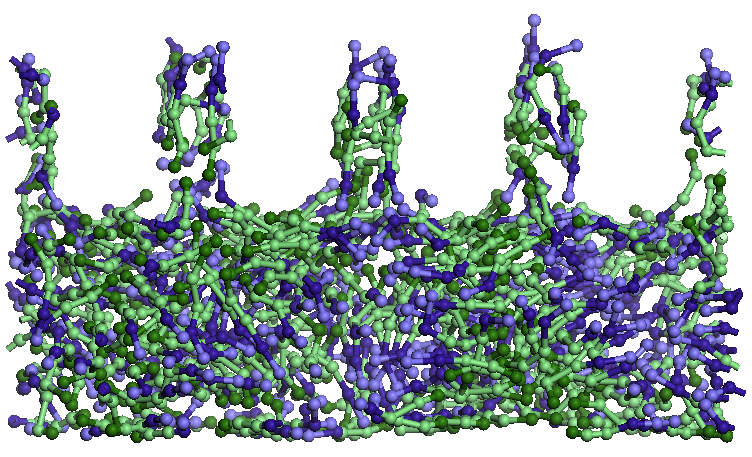

Figura 7.
Detalii ale structurilor formate de moleculele de apa (sfere verzi
izolate) si
SDS (lanturi, in care capetele albastre reprezinta ionii de sodiu)
localizate
in apropierea carbonului amorf (stanga) si respectiv adsorbite pe
stratul
superior de grafena (dreapta).
Pe suprafata grafenei
superioare un numar redus de molecule de apa si de SDS adsorb, formand
un
strat. Moleculele SDS adsorb paralel cu stratul de grafena, o singura
molecula
fiind perpendiculara pe strat. Cateva molecule de apa si SDS intra si
ies din
pori sau isi modifica orientarea. Dimensiunea porilor este suficient de
mare
pentru a avea acces 4-5 molecule SDS si cateva molecule de apa.
Moleculele de
apa sunt fortate sa paraseasca porii la scurt timp (1-2 ps) dupa ce
patrund in
ei. Moleculele SDS ce patrund in pori au o orientare cu capatul
hidrofil catre
partea superioara a porilor, ramanand cu capatul hidrofobic orientat
catre
solutie. Stratul de solutia apa-SDS isi modifica structura initiala
prin
rearanjarea moleculelor SDS in solutie: moleculele de sub pori se
orienteaza
vertical cu capatul hidrofob spre por, avand in vecinatate capetele
hidrofobe
ale moleculelor SDS intrate in pori iar restul moleculelor SDS se
orienteza cu
capetele hidrofobe catre capetele hidrofile ale moleculelor SDS din
apropierea
porilor. Moleculele SDS din zona inferioara a stratului solutiei se
orienteaza
aproape orizontal incercand sa aiba capetele orientate catre zone de
acelasi
tip. Este de presupus ca in aceasta restructurare dimensiunea, forma si
spatierea porilor joaca un rol important la fel ca si temperatura,
grosimea
stratului de solutie si concentratia SDS.
|
|
|
|
|
0.05
ns |
0.10
ns |
50.00
ns |


Figura 7. Evolutia
sistemului in
timpul simularilor de dinamica moleculara, in conditiile impunerii unui
sistem
poros flexibil, pentru pori cu dimensiunea medie de 13 A.
In cazul flexibilizarii complete a structurilor de carbon evolutia
sistemului in primele 50 ps este
foarte similara cu cea din cazul simularii cu structurile de carbon
rigide dar
dupa alte 10 ps cele doua straturi de grafit se onduleaza datorita
vibratiilor
iar carbonul poros se extinde pe verticala cu aproximativ 2-3%. In
decursul a
catorva nanosecunde solutia apa-SDS este constransa intre cele deoua
structuri
de carbon. Aici poate fi vorba de unele artefacte in cea ce priveste
conditiile
initiale ale simularii, respectiv alegerea aleatoare a vitezelor
initiale ale
atomilor de carbon pot da acestor sisteme miscari toatale de rotatie
care in
conditiile ‚frecarii‘ intre imaginile straturilor de carbon sa
transforme
aceste miscari de rotatie in vibratii interne si de aici sa avem aceste
ondulari excesive ale straturilor de grafena. Totusi aceste artefacte
nu au o
influenta esentiala asupra moleculeor patrunse in pori.